Keloid Scarring: Understanding the Genetic Basis, Advances, and Prospects
Article information
Abstract
Keloid disease is a fibroproliferative dermal tumor with an unknown etiology that occurs after a skin injury in genetically susceptible individuals. Increased familial aggregation, a higher prevalence in certain races, parallelism in identical twins, and alteration in gene expression all favor a remarkable genetic contribution to keloid pathology. It seems that the environment triggers the disease in genetically susceptible individuals. Several genes have been implicated in the etiology of keloid disease, but no single gene mutation has thus far been found to be responsible. Therefore, a combination of methods such as association, gene-gene interaction, epigenetics, linkage, gene expression, and protein analysis should be applied to determine keloid etiology.
INTRODUCTION
The skin is the largest organ and protects against dangerous environmental factors and water loss. Skin is composed of two main layers: the dermis and the epidermis. The dermis contains a variable amount of fat, collagen, and elastic fibers that provide strength and flexibility to the skin.
Keloids are benign hyperproliferative growths of dermal fibroblasts characterized by the excessive deposition of extracellular matrix components, especially collagen, fibronectin, elastin, proteoglycans, and growth factors such as transforming growth factor (TGF) β [1]. The mechanisms of keloid formation include alterations in growth factors, collagen turnover, and tension alignment, as well as genetic and immunological contributions. Trauma, foreign-body reactions, infections, and endocrine dysfunctions have all been proposed as risk factors for the development of keloids after surgery in genetically susceptible people [1,2]. Keloid disease affects both sexes equally [3,4] and can occur at every age but is more prevalent between the ages of 10 and 30 [5].
EVIDENCE OF A GENETIC BASIS FOR KELOID DISEASE
Evidence from ethnic groups, families, and twins
The incidence of keloids is different among populations that reflect different etiologic factors. The worldwide keloid disease prevalence varies according to ethnicity. Patients with darker skin, however, have a higher prevalence than those with lighter pigmentation [6]. It is the fifth most common skin disease in adult black patients in the United Kingdom [3] and the most common skin disease among ethnic Chinese patients in Asia [7]. In addition, many reports have been published on familial keloid cases [5,8-11], reflecting the importance of genetic factors among these families. Finally, the high frequency of identical twins both developing keloids also strongly supports a role for genetics in keloid etiology [5,12].
Both autosomal dominant with incomplete penetrance and variable expression modes [5,9,10] as well as autosomal recessive modes of inheritance [13] have been seen among families with keloid disease. Clinical and genetic heterogeneity with a variable clinical expressivity between families and within the affected members of same family has been noted [5,8-11]. While the autosomal dominant mode of inheritance with incomplete penetrance is the most common reported model for keloid disease, it remains unclear whether keloid disease is a simple Mendelian or a complex oligogenic disorder. As reviewed more recently, it is well known that environmental factors can trigger the formation of keloids in genetically susceptibility individuals [14]. In addition, studies on people of different ethnicities have discovered non-overlapping associated genes and genomic regions. Taken together, these investigations indicate that a complex inheritance model with contributions from multiple genetic factors along with triggering environmental influences could be the best model for keloid disease. More specifically, it seems plausible that an autosomal gene may play a major role in combination with more moderate recessive gene effects.
Evidence from Mendelian disorders with keloid symptoms
Several Mendelian disorders manifest keloids as part of their clinical features. Individuals with a connective-tissue disorder, for example, have a possibility of developing keloids as part of their disease. Almost all Mendelian syndromic forms of keloid disease such as lateral meningocele (OMIM #130720), Rubinstein-Taybi (OMIM #180849), Leigh necrotizing encephalomyelopathy (OMIM #161700), Ullrich congenital muscular dystrophy (UCMD; OMIM #254090), Ehlers-Danlos syndrome (OMIM #130050), and Goeminne TKCR syndrome (OMIM #314300) have shown a dominant mode of inheritance that is consistent with the mode of inheritance among families with non-syndromic keloid disease [5,9,10].
Rubinstein-Taybi syndrome 1 (RSTS1) patients develop keloids with high frequency. RSTS1 is caused by a contiguous gene deletion involving the CREBBP gene as well as other neighboring genes on the chromosome 16p13.3 (OMIM #180849). A questionnaire-based study of 61 adults with RSTS ranging in age from 18 to 67 years found that 57% of patients developed keloids [15]. In addition, 28 patients exhibited keloids in a series of 574 examined individuals with RSTS [16]. The high incidence of keloids as both a proliferative disorder and as neoplasms in RSTS patients is attributed to the function of CREBBP in cAMP-regulated cell immortalization [17].
Nadeau et al. [18] reported the medical history of 13 patients with UCMD, three of which also had keloids. UCMD is a heterogeneous disease mainly caused by mutations in collagen genes. Similarly, Ehlers-Danlos syndrome (EDS) is an autosomal dominant connective-tissue disorder that manifests keloids as one of the clinical symptoms. EDS type IV is caused by a heterozygous mutation in the gene for type III collagen (COL3A1; 120180) on chromosome 2q31, close to the gene locus for UCMD syndrome. In addition, overlapping phenotypes have been observed between EDS and UCMD. Finally, Goeminne TKCR syndrome was first reported in a family with six affected members in whom two patients also developed multiple keloids [19].
Therefore, familial aggregation, occurrence in identical twins, Mendelian modes of inheritance, expression studies, and the high prevalence of keloids among different ancestries all provide strong evidence in favor of genetic factors in keloid formation.
GENETIC STUDIES OF KELOID DISEASE
Evidence from expression studies and gene interaction
There are 166 studies on PubMed with "gene expression in keloid" as a keyword. With the advent of microarray expression platforms, a long list of genes has been found to be either up-regulated or down-regulated in keloid samples. Keloid disease is a complex condition in which multiple interactions between the susceptible genes and their products have been reported. Keloids are enriched in growth factors and extracellular matrix (ECM) molecules, and fibroblasts compose the majority of dermis cells responsible for the production and remodeling of extracellular matrix during wound healing. ECM molecules play an important role in skin structure; therefore, disruption of the ECM could be responsible for abnormal scar tissue formation [20].
As shown in Fig. 1, collagens (type I and III), microfibrillar proteins (elastin, fibrillin) and hyaluronic acid are three major structural elements of the ECM [20]. Of these, collagens act as a network throughout the dermis to maintain tissue integrity and microfibrillar proteins allow flexibility in the deeper dermis [20]. Extensive expression studies focusing on ECM proteins have been carried out. Most expression studies have been conducted on keloid-derived cultured fibroblast cells in comparison with normal tissue. The influence of cell culture components, which are in direct interaction with fibroblast cells, on gene expression is unavoidable. Keloid tissue is characterized by the accumulation of extracellular matrix, especially collagen (Fig. 1). In addition, keloid-derived fibroblast cells exhibit high expression of TGF β1 and TGF β2 [21] and consequently over-synthesis of collagen [22,23]. TGF β stimulates the synthesis of collagen and promotes wound healing via regulating the growth, differentiation, and proliferation of fibroblast cells [24]. TGF β3 stimulates collagen synthesis via TGF β1 [22] and TGF β2 [23]. In addition, the TGF β pathway is involved in fibrosis as well as several other fibrotic disorders [14]. It has been shown that in the primary stages of fibrosis in keloid tissues, TGF β1 is expressed by neovascular endothelial cells, which consequently stimulates the expression of type I and VI collagens at a high level [25]. TGF β1 is a key factor in keloid development and regulates the expression of multiple downstream genes. Exogenous TGF β1 upregulates the expression of platelet derived growth factor (PDGF) α receptor in keloid-derived fibroblast cells but not in non-keloid-derived fibroblast cells [26]. It has also been shown that TGF β stimulates the expression of vascular endothelial growth factor (VEGF) in keloid fibroblasts as well [27].
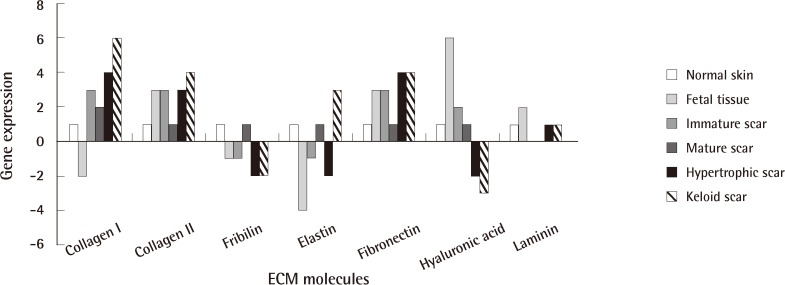
Extracellular matrix (ECM) in normal skin, and hypertrophic and keloid tissue
ECM components in normal skin compared with hypertrophic scars and keloid tissue. The numbers indicate relative expression. Six means very high expression and -4 indicates very low expression.
SMAD genes act downstream of TGF β in keloid development. In keloid fibroblast cells, SMAD2 small interfering RNAs (siRNA) caused the downregulation of SMAD2 and SMAD3, resulting in a decrease in procollagen levels [28,29]. This indicates that SMAD2 and SMAD3 play a critical role in TGF β-induced fibrosis in the formation of keloids.
EVIDENCE FROM LINKAGE AND ASSOCIATION STUDIES
As mentioned above, familial aggregation studies shed light on the genetic risk factors that might be responsible for keloid formation. Several researchers have since attempted to map the susceptible locus (or loci) of keloid disease in these families.
Genome-wide linkage studies on a Japanese and an African-American family with keloid disease resulted in the detection of linkage intervals on chromosomal regions 2q23 and 7p11, respectively [9]. However, linkage to 7p11 was excluded from a large Chinese family with keloid disease [30], and linkage intervals at 15q22.31-q23, 18q21.1, and 10q23.31 were found for this family [31,32]. The 18q21.1 region harbors the SMAD 2, 7, and 4 genes, which are involved in regulating of TGF β signaling pathway.
Multiple case-control studies have been published on the associations between keloid disease and keloid candidate genes [33-40]. Despite strong evidence from expression studies, no study has yet found any association between TGF β family members and keloid disease in Caucasian populations [33-36]. However, TGF β1 plasma concentration was found to be associated with the -509 T>C variant of TGF β1 [41], conflicting with results observed in another study [34].
In addition, downstream genes in the TGF β signaling pathway, namely SMAD 3, 6, and 7, did not attain statistical significance in a case-control study examining keloid disease in the Afro-Caribbean ethnicity [42].
In contrast to TGF β family members, HLA genes have attained statistical significance in several studies [37,39,43]. Of these, HLA-DRB1*15 appeared to be the most robust with replication in both Chinese and Caucasian ethnic groups [37,39], suggesting that, at least in these ethnic groups, HLA-DRB1*15 might be associated with an increased risk of keloid disease.
GENOME-WIDE STUDIES IN KELOID DISEASE
With the advent of high-throughput microarray genotyping technologies, researchers have used these methods to look through the entire genome. However, there is still no genome-wide linkage study using high-density single-nucleotide polymorphism (SNP) map arrays on families with keloid disease. Recently, however, in a genome-wide case-control association study on a Japanese population, four susceptibility loci for keloid disease were detected including 1q41, 3q22.3-23, and 15q21.3 [44]. A replication study confirmed the possible role of the NEDD4 gene in the 15q21.3 chromosomal region. It has since been shown that NEDD4 upregulates fibronectin and type 1 collagen and so plays a role in the accumulation of extracellular matrix [45]. In another study, copy number variations in 6p21.32, 11q11, 17q12, 8p23.1, 22q13.1, 19p13.1, and 2q14.3 were detected using an array-based comparative genomic hybridization [43]. Region 6p21.32, which harbors HLA-DRB5, also showed a significant association in the validation study.
THE ROLE OF EPIGENETICS IN KELOID DISEASE
The role of epigenetics in the formation of malignancies is well known. As a keloid is a benign tumor, the possibility of epigenetic alterations in keloid tissue exists. Indeed, it was recently reported that keloid fibroblast cells have altered patterns of DNA methylation and histone acetylation [46]. Examining the methylation profile could give new insight into keloid treatment. However, additional study is needed to fully address the role of epigenetics in the etiology of keloid disease.
CONCLUDING REMARKS AND OUTLOOK
Keloid disease is a complex condition in which different ethnic groups show different susceptibilities to the development of the disease. The differences in prevalence between populations probably reflect the contributions of different genetic risk factors. In general, most genes in complex disorders have a moderate effect; therefore, association studies, which are powerful methods for detecting genes with moderate effects, are excellent for examining these types of disorders. A range of statistical methods including single marker analysis, genotype analysis, haplotype analysis, relative risk analysis, and gene-gene and gene-environment interaction analyses are also needed to find candidate genes in keloid disease etiology. In addition, a lack of reproducibility among some populations might be the result of small sample size, population stratification, inappropriate statistical analysis methods, or real heterogeneity among populations.
A change in an amino acid alone may not be a sufficient criterion for predicting disease in complex traits such as keloid. Variants could also exert effects by disrupting or activating the function of splicing binding sites, or hypomorphic variants could exert their effects at the transcription level and interact with other risk factors. However, as no single mutation inside a gene has been detected so far, a multifactorial inheritance model would fit best. Therefore, the interaction between genes and environmental factors, as well as the possible role of coding and regulatory variants and epigenetics must be accounted for in future studies.
Notes
This study was supported by a Universiti Sains Malaysia (USM) short-term grant (No. 304/PPSP/61310017).
No potential conflict of interest relevant to this article was reported.