The molecular pathophysiology of vascular anomalies: Genomic research
Article information

Abstract
Vascular anomalies are congenital localized abnormalities that result from improper development and maintenance of the vasculature. The lesions of vascular anomalies vary in location, type, and clinical severity of the phenotype, and the current treatment options are often unsatisfactory. Most vascular anomalies are sporadic, but patterns of inheritance have been noted in some cases, making genetic analysis relevant. Developments in the field of genomics, including next-generation sequencing, have provided novel insights into the genetic and molecular pathophysiological mechanisms underlying vascular anomalies. These insights may pave the way for new approaches to molecular diagnosis and potential disease-specific therapies. This article provides an introduction to genetic testing for vascular anomalies and presents a brief summary of the etiology and genetics of vascular anomalies.
INTRODUCTION
The clinicopathological classification of vascular anomalies, which was first proposed by Mulliken and Glowacki in 1982 [1], has continued to be a basic standard. The classification of these anomalies into two broad categories—tumors and vascular malformations—was the basis of the 2018 classification proposed by the International Society for the Study of Vascular Anomalies (ISSVA). However, there are still many limitations of pathophysiological analyses on the basis of clinical and histological findings [2-4]. Since recent studies have identified many genes potentially linked to vascular anomalies, several pathophysiological analyses based on these genes have been conducted, and potential new treatments have also been proposed. Since the 1990s, the genetic basis for vascular anomalies has been elucidated, and as a result, mutations associated with numerous types of lesions have been reported over the past few years [3]. Recent developments in sequencing technologies have opened new horizons in terms of genomic studies, and research in this field is actively expanding [5]. This article presents an introduction to genetic testing for vascular anomalies and a brief summary of the etiology and genetics of vascular anomalies.
GENETIC TESTS FOR VASCULAR ANOMALIES
In the past few decades, remarkable and unexpected advances in genetic testing have been made. The completion of the Human Genome Project in 2003 was followed by a veritable flood of discoveries regarding the genetic basis of various diseases [6]. Eventually, patients’ genomes will be sequenced to identify genetic diseases, to analyze the causes of complex multifactorial diseases, and to predict the best treatment and prognosis for each individual.
There are two major classes of genetic variations: copy number variations (CNVs) and single-nucleotide variants (SNVs). A CNV is defined as a change in length of 1,000 or more base sequences. Genome-wide CNVs can be detected using a chromosomal microarray (CMA) with high resolution. SNVs take place when a single nucleotide (e.g., A, T, C, or G) is altered in the DNA sequence. SNVs are the most frequent genomic variations.
Sequencing refers to reading the DNA base sequence containing genetic information in order. DNA sequencing can be broadly divided into Sanger sequencing and next-generation sequencing (NGS) [7]. The Sanger method involves sequencing single strands of DNA, whereas in NGS, one long DNA strand is cut into millions of strands, and then genes are amplified and sequenced at once [7,8]. NGS is composed of four basic steps. The first step is library preparation, in which extracted DNA is cut into millions of pieces randomly and then adapters are attached to the 5´ and 3´ ends of DNA for further polymerase chain reaction (PCR). Second, in the cluster generation step, each of the millions of DNA fragments is amplified through PCR to create clonal clusters. Third, in the sequencing step, complementary nucleotides are generated by DNA polymerase from the DNA strand cluster and the nucleotide type is identified with fluorescence. Fourth, in the data analysis step, the sequence data of millions of short reads are arranged using the reference genome and sequencing results are obtained through bioinformatics. Sanger sequencing reads hundreds of nucleotide sequences in detail, whereas NGS can quickly identify sequences across genomes at a low cost. The general category of NGS includes whole exome sequencing (WES), targeted gene panel sequencing (TS), and whole genome sequencing (WGS).
Applicable genetic testing methods for vascular anomalies are Sanger sequencing for a defined phenotype with a monogenic disorder, CMA, TS, WES, and WGS. The testing method should be carefully chosen according to the purpose of testing. Usually, CNVs are analyzed by CMA or WGS, whereas SNVs and small insertions/deletions are more suitable for analysis by sequencing, WES, or WGS. Genetic testing can follow an orderly progression if the diagnosis remains unclear.
Mutations can be classified into germline mutations and somatic mutations. Germline mutations pass from the parent to the offspring through germ cells, and can therefore be found in almost every cell in an individual. Somatic mutations occur only in a subgroup of cells (excluding germ cells) in an individual. Therefore, these mutations are not transmitted to the next generation. Instead, they are only transmitted to the offspring of the affected cells, giving rise to somatic mosaicism, which plays a major role in the development of vascular lesions. Somatic mutations can cause disease, as exemplified by cancer, where cells acquire somatic mutations and develop into tumors.
Although familial inheritance has also been reported, most vascular anomalies are sporadic. Germline mutations can be tested using blood, buccal mucosal cells, or lesion tissue samples; however, somatic mutations can only be found in lesion tissue samples. NGS has proven to be a valuable tool for discovering somatic mutations [9,10].
GENETICS OF VASCULAR ANOMALIES
The causal genes of vascular anomalies known to date are well organized in the ISSVA classification of vascular anomalies (c2018 ISSVA, available from: issva.org/classification) [11].
Vascular tumors
Infantile hemangioma
Infantile hemangioma (IH) arises from the proliferation of endothelial cells (ECs). Two main proposals have been made regarding the origins of ECs in IH: embolic placental angioblasts, which share placental markers (glucose transporter protein 1 [GLUT1], Fcγ receptor II, Lewis Y antigen, and merosin), and endothelial progenitor cells (CD133+/CD34+ circulating progenitor cells and stem cells) [12,13]. Vascular endothelial growth factor (VEGF)-A signaling is associated with IH. In the ECs in IH, the formation of IH has been shown to be caused by changes in the VEGF-A signaling pathway that are related to missense mutations in the genes encoding VEGFR2 (KDR) and TEM8 (ANTXR1) [14,15].
Congenital hemangioma
There are three types of congenital hemangioma (CH) that appear fully formed at birth: non-involuting CH, partially involuting CH, and rapidly involuting CH [15,16]. They are all GLUT1-negative. It has been reported that there is a mutation at the glutamine 209 (Gln209) position in GNAQ or GNA11, and the Gln209 missense mutation is known to activate GTPdependent signaling, which leads to the constitutive activation of MAPK and/or YAP signaling [17].
Pyogenic granuloma
Pyogenic granuloma (PG) may be isolated or associated with a capillary malformation (CM). It has been reported that PGs associated with CM (secondary PGs) show a somatic GNAQ mutation reflecting an origin from CM cells. Furthermore, BRAF somatic mutations were found in eight of 10 secondary PGs and NRAS mutations in one of 10. However, isolated PGs were associated with BRAF (four of 25) or KRAS (one of 25) mutations [18]. A somatic activating GNA14 mutation has been reported in one case and a GNA11 mutation in two cases [19].
Kaposiform hemangioendothelioma
Kaposiform hemangioendothelioma (KHE) is usually present at birth and is typically diagnosed in infancy or early childhood. The incidence of the Kasabach-Merritt phenomenon has been estimated at 42% to 71% in KHE [20]. GNA14 mutations were found in one of three KHE specimens and one of four closely related tufted angiomas [19].
Vascular malformations
Capillary malformation
CM is the most common type of vascular malformation [21]. GNAQ mutations were found in 80%–90% of cases with a sporadic or syndromic (Sturge-Weber syndrome) etiology [22]. GNA11 mutations were identified in three of eight specimens with diffuse CM and overgrowth of an extremity [23]. The GNAQ mutations in CM primarily present in ECs [24]. Atypical CM may co-occur with arteriovenous malformation (AVM) in a single patient. This entity, which is referred to as CM-AVM, can be subdivided into CM-AVM1 and CM-AVM2. CM-AVM1 is inherited in an autosomal dominant pattern and is caused by a RASA1 mutation [25]. One-third of patients with CM-AVM1 have fast-flow lesions; in contrast, the risk of fast-flow malformations is lower in CM-AVM2, which is caused by a loss-of-function mutation in EPHB4 [26].
Lymphatic malformations
Lymphatic malformations (LMs) are sporadic slow-flow lesions that are composed of multiple cystic structures, which can be macroscopic, microscopic or combined [27]. PIK3CA mutations were found in 16 of 17 specimens. Mutations in the PIK3CA gene can enhance the ability of its protein product to bind to the cell membrane or activate its kinase, which results in activation of the AKT/mTOR signaling pathway [28,29]. Rapamycin (sirolimus), which is well known as an mTOR inhibitor, has shown good efficacy and a favorable safety profile when used in patients [10]. Primary lymphedema is a type of vascular malformation that can be hereditary. Several mutations (VEGFR3/ FLT-4, FOXC2, SOX18, CCBE1, etc.) have been identified in patients with primary lymphedema [15].
Venous malformations
Venous malformations (VMs) are usually sporadic, but can be familial [30]. Cutaneomucosal autosomal dominant VM was found to be caused by TIE2 mutations, which were found in 80 of 130 specimens of sporadic VMs. Activating somatic mutations of PIK3CA have also been identified in sporadic VMs that lacked TEK/TIE2 mutations [3,15,31]. Verrucous VMs associated with the MAP3K3 mutation are hyperkeratotic abnormalities that affect the skin of limbs [32]. Glomuvenous malformation (GVM) is an autosomal dominant disease in which several small lesions are caused by germline or somatic loss-of-function mutations in glomulin (GLMN) [33]. GVMs have reported as requiring somatic second-hit mutations to trigger disease onset; the first mutation is inherited in an autosomal dominant manner, while the second mutation is acquired. Blue rubber bleb nevus syndrome, which is a non-hereditary condition, presents as multifocal VMs. This syndrome is associated with TIE2 mutations [34]. Cerebral cavernous malformations (CCMs) are characterized by enlarged capillary cavities in the central nervous system. In 9% of CCM patients, cutaneous lesions are also found. CCMs, which are associated with mutations in CCM1/KRIT1, CCM2/malcavernin, and CCM3/PDCD10, can be sporadic or familial [5,35].
Arteriovenous malformations
AVMs are anomalous connections between arteries and veins through a nidus or fistula, bypassing high-resistance capillary beds [36]. AVMs occur sporadically, and are associated with a mutation in the MAP2K1 gene [37]. Inherited AVMs can occur in the context of other conditions, such as hereditary hemorrhagic telangiectasia (HHT), and combined diseases such as CM-AVM (see CM) and Parkes Weber syndrome [10,15,24].
Hereditary hemorrhagic telangiectasia
HHT is an autosomal dominant vascular dysplasia that can cause epistaxis, mucocutaneous telangiectasias, and/or visceral AVMs. Ninety percent of HHT cases are associated with a lossof-function mutation of one of three identified genes. HHT1 is associated with mutations in endoglin (ENG), a co-receptor for activin-like receptor; HHT2 with mutations in activin receptorlike kinase 1 (ACVRL1); and juvenile polyposis/HHT syndrome with mutations in MADH4, which encodes the downstream effector SMAD4. HHT is associated with germline mutations in genes involved in the TGF-β/BMP signaling pathway [5,38,39].
Vascular malformations associated with other anomalies
Vascular malformations are one of the major components in various syndromes that cause enlargement of soft tissues or bones. Klippel-Trenaunay syndrome (KTS) presents with a classic triad of port-wine stains, asymmetric extremity overgrowth, and underlying VMs or LMs [40,41]. In 19 of 21 KTS patients, PIK3CA mutations were found to play a role in the pathogenesis of the condition [42]. Macrocephaly-CM usually causes neurological abnormalities, and patients typically have CM on the upper lip, back, and/or limbs. These patients also have PIK3CA mutations [43]. CLOVES syndrome is characterized by congenital lipomatosis, overgrowth, vascular malformations, epidermal nevi, and skeletal anomalies. Mutations in PIK3CA were identified in 36 of 38 patients [44]. Proteus syndrome, which features asymmetric growth of skeletal and connective tissue, cerebriform nevi of the hands or feet, epidermal nevi, and underlying vascular malformations, results from a somatic activating mutation in AKT1 [45]. Maffucci syndrome features several enchondromas and soft-tissue VMs. This condition results from somatic mutations of isocitrate dehydrogenase (IDH), with 98% of cases caused by IDH1 mutations and 2% by IDH2 mutations [46].
ABNORMAL SIGNALING PATHWAYS IN VASCULAR ANOMALIES
These genetic mutations directly change the activities of intracellular signaling pathways, thereby influencing various downstream actions. The pathways most involved in vascular anomalies are the PI3K/AKT/mTOR and RAS/MAPK signaling pathways. Another important signaling mechanism is the TGF-β/SMAD signaling pathway, which plays a role in HHT (Fig. 1).
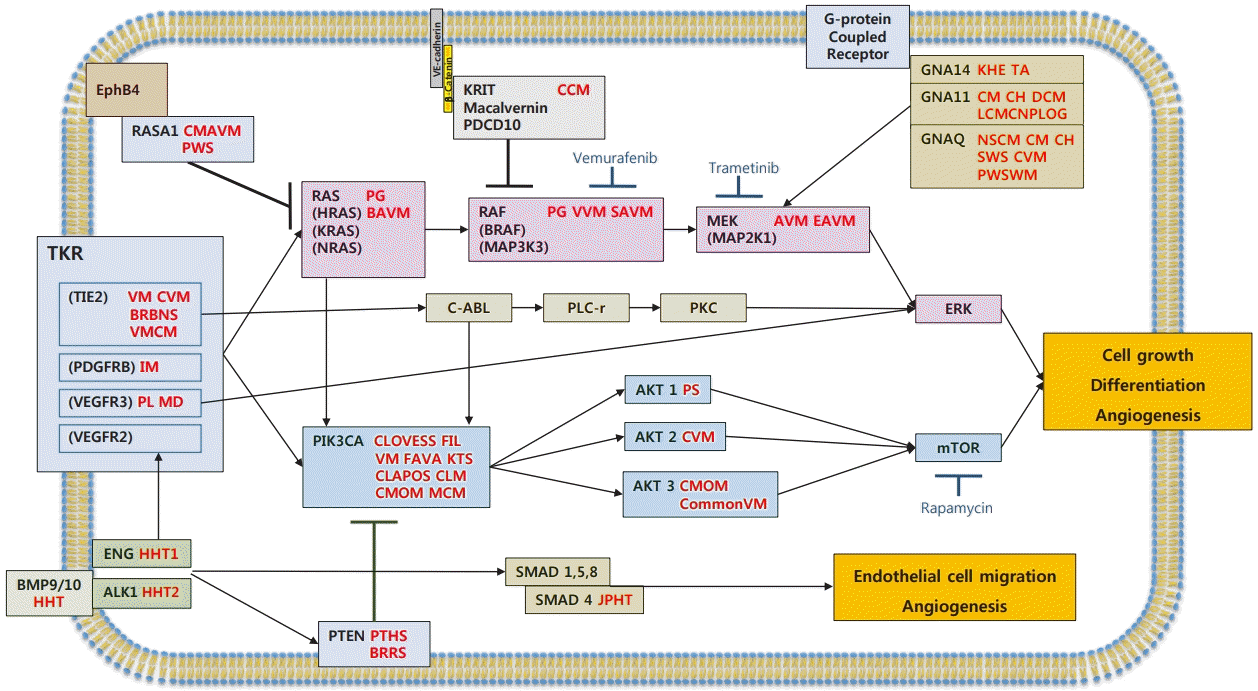
Mutations and signaling pathways in vascular anomalies
Mutations in vascular abnormalities affect genes involved in tyrosine kinase signaling via the RAS or PIK3CA pathways. This figure is a schematic diagram of key genetic mutations related to signal transduction and vascular malformations in endothelial cells. Proteins mutated in other vascular disorders are indicated. Mutations in GNAQ/GNA11/GNA14, RASA1, and KRIT lead to constitutive activation of RAS/RAF/MEK/ERK signaling. Mutations in TIE2/TEK lead to permanent activation of the PIK3CA/AKT/mTOR pathway. AVM, arteriovenous malformation; BAVM, brain arteriovenous malformation; BRBNS, blue rubber bleb nevus syndrome; BRRS, Bannayan-Riley-Ruvalcaba syndrome; CCM, cerebral cavernous malformation; CH, congenital hemangioma; CLAPOS, CLAPO syndrome; CLM, cystic lymphatic malformation; CLOVESS, CLOVES syndrome; CM, capillary malformation; CommonVM, common venous malformation; CMAVM, capillary malformation–arteriovenous malformation 1,2; CMOM, capillary malformation of macrocephaly; CVM, cutaneomucosal venous malformation; CH DCM, diffuse capillary malformation; EAVM, extracranial arteriovenous malformation; FIL, facial infiltrating lipomatosis; FAVA, fibroadipose vascular anomaly; VMCM, Familial venous malformation cutaneous and mucosal; HHT, hereditary hemorrhagic telangiectasia; IM, infantile myofibroma; JPHT, juvenile polyposis hemorrhagic telangiectasia; KHE, kaposiform hemangioendothelioma; KTS, Klippel-Trenaunay syndrome; LCMCNPLOG, limb capillary malformation with congenital nonprogressive limb overgrowth; MD, Milroy’s disease; MCM, megalencephaly capillary malformation; NSCM, non-syndromic capillary malformation; PL, primary lymphedema; PS, Proteus syndrome; PTHS, PTEN hamartoma syndrome; PG, pyogenic granuloma; PWS, Parkes Weber syndrome; PWSWM, port wine stain with macrocheilia; SVAM, spinal arteriovenous malformation; SWS, Surge-Weber syndrome; TA, tufted angioma; VM, venous malformation; VVM, verrucous venous malformation.
CONCLUSIONS
Advances in DNA sequencing have yielded extensive knowledge regarding the etiological molecular mechanisms involved in the development of vascular anomalies. NGS is a very effective method of analyzing germline and somatic mutations of vascular anomalies, which have various phenotypes, and their associated dysfunctions. An important point to be emphasized is that somatic mutations and their mechanisms for vascular anomalies, which mainly are sporadic and unifocal, should be further studied in the future. Although many downstream effects remain unknown, it is clear that normalization of the affected signaling pathways is an important target for treatment. Mutation-specific targeted therapies are also being studied, which may result in a paradigm shift in therapeutic approaches to vascular anomalies. Further research into the cellular effects of mutations is expected to yield insights into the underlying pathophysiology that will enable new therapies to be developed.
Notes
Conflict of interest
No potential conflict of interest relevant to this article was reported.
Author contribution
Conceptualization: JS Kim, SK Hwang, HY Chung. Data curation: JS Kim, SK Hwang, HY Chung. Formal analysis: HY Chung. Funding acquisition: HY Chung. Methodology: HY Chung. Project administration: HY Chung. Visualization: JS Kim, HY Chung. Writing - original draft: JS Kim, HY Chung. Writing - review & editing: JS Kim, SK Hwang, HY Chung.