Bart Syndrome
Article information
Bart syndrome is a rare inherited disorder characterized by the localized absence of the skin, blister formation, and nail deformity and is considered a variant of aplasia cutis congenita (ACC) with epidermolysis bullosa (EB) [1]. Bart et al. [1] introduced this syndrome in a 1966 report describing a single kinship with a combination of these traits. To the best of our knowledge there are very few cases of Bart syndrome reported in the PubMed database. Here, we present a newborn female baby with Bart syndrome involving ACC in the lower extremities and right hand as well as EB in the right hand and right lower extremity. A female infant was born during the 40th week of the mother's third pregnancy. The patient was referred to the newborn intensive care unit of our hospital, which consulted with the department of plastic surgery because of a skin problem. On physical examination, the patient had extensive absence of skin on the right hand and both lower extremities, a single bullous lesion on the right hand, and three bullous lesions on the right lower extremity. The skin lesion was enveloped in an ultrathin translucent membrane, and there were some blisters on the membrane. Moreover, the patient had a nail deformity of great toe (Fig. 1). The patient's mother had an absence of skin at birth as evidenced by a thin but broad scar on her left shin. Except for the mother's history, there was no other specific family history. A biopsy of the bullous lesion on the edge of the right foot was performed. Light microscopic examination revealed a subepidermal blister, and electron microscopy showed evidence of separation of the dermal-epidermal junction and interruption of the basal lamina, similar to what is seen in junctional EB (Fig. 2).
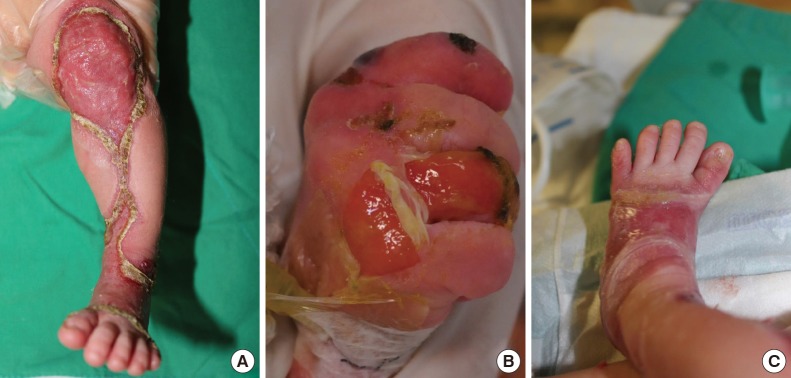
Pretreatment photograph. (A) Extensive absence of skin on the left lower extremity, (B) a ruptured blister on the right hand, and (C) nail deformity of the left great toe.
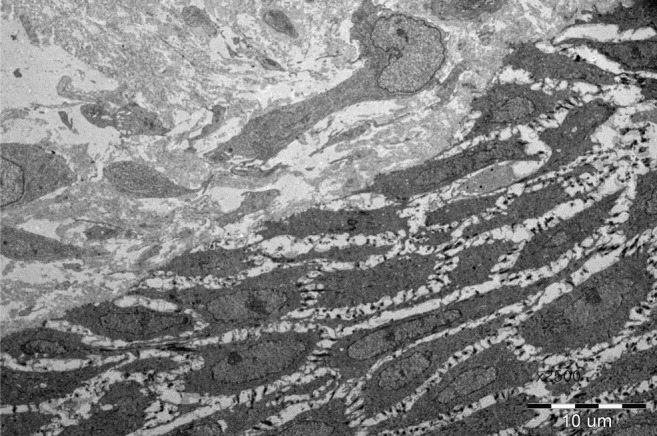
Histopathologic findings of the epidermolysis bullosa. Electron microscopic finding showing a widened dermal-epidermal junction and interruption of the basal lamina.
The patient was managed conservatively without operation. When the patient was referred to our hospital, there were crusts and dead tissues on wounds without infection sign. We cleaned crusts and dead tissues using mechanical debridement and hydrodebridement with normal saline and applied antibacterial ointment to protect from infection and to induce wound contraction. Petrolatum gauze dressing was also applied on wounds for maintaining hydration. Intravenous or oral antibiotics were not used. After three months, the skin lesions healed completely with epithelialization with only a pigmented scars remaining in place of the lesions (Fig. 3). Bart syndrome was first described by Bart et al. [1], who reported a kinship consisting of 103 direct descendants from one proband over four subsequent generations. Among the members of this kinship, 25 members had one or more of three traits distinct traits, namely, congenital localized absence of skin involving the lower extremities, mucocutaneous blistering, and nail abnormalities. Following this report, the combination of ACC affecting the lower extremities with EB and nail abnormalities has been referred to as Bart syndrome [1]. Bart syndrome is characterized by a combination of ACC and EB features [1]. ACC is a rare congenital anomaly characterized by an absence of skin [2] and was first described in 1767 by Cordon. ACC is classified into nine groups based on the location and presence of other abnormalities as described by Frieden (Table 1) [3]. ACC occurs most commonly on the scalp and cranial vault; however, it may affect any location on the body [2]. The depth of ACC lesions can be quite diverse. At birth, skin defects associated with ACC can range from superficial erosion to complete absence of all skin layers [2]. EB is a rare inherited disorder characterized by blister formation due to increased skin and mucous membranes fragility [4,5]. In general, four major types of EB have been described according to the level of skin cleavage at the ultrastructural level. EB can be further classified into more than 30 subtypes based on the pattern of inheritance, clinical findings, and molecular defects (Table 2) [4,5]. The diagnosis of EB is based on medical and family history of the patient. However, it can difficult to make a precise diagnosis without laboratory tests. Therefore, skin biopsy with immunofluorescent mapping (IFM) and/or electron microscopy (EM) are needed to definitively diagnoses and classify the type and subtype of EB [5]. IFM is the primary test for definitive diagnosis of EB, since IFM is considered to have a higher sensitivity and specificity than EM [4,5]. However, in cases where a satisfactory diagnosis cannot be made despite repeated IFM analysis, EM can be useful [4,5] as it allows for characterization of the ultra-structure of skin at the level of dermal-epidermal junctions [4]. Genetic testing is the most accurate method for analyzing EB and can provide a definitive diagnosis of EB type and subtype through precise determination of the location and type of mutation. However, genetic testing is more expensive than IFM and EM, and thus its use is limited [4,5]. Management of Bart syndrome ranges from conservative to secondary intention, to surgical intervention [1,3,4]. Conservative management involves local wound care and infection control using systemic antibiotics. Specifically, after determining the location, size, type, and severity of the each wound, they are cleansed by normal saline or diluted antiseptics [4]. When blister formation on wounds is noted, the blister fluid may be removed by sterile needle [5]. If needed, hydrodebridement and mechanical debridement can be performed to prevent infection and help wound contraction [4]. Topical agents such as silver sulfadiazine cream or bacitracin ointment can also be applied to wounds [3]. For dressing, Petrolatum gauze seems to be the most effective dressing material for keeping wounds moist, protecting the site from trauma, and lowering the risk of skin problem such as contact dermatitis [3,4]. Administration of systemic antibiotics is not routine, but can be used if infection is suspected [4]. While most patients can be treated with conservative management, deep and large wounds may require surgical intervention such as skin grafting or local flaps [2,3]. In this report, we present a rare case of Bart syndrome in a newborn. Several treatment options are available for this condition according to severity; however, it is important to note that most cases can be managed by conservative therapy without surgical intervention.
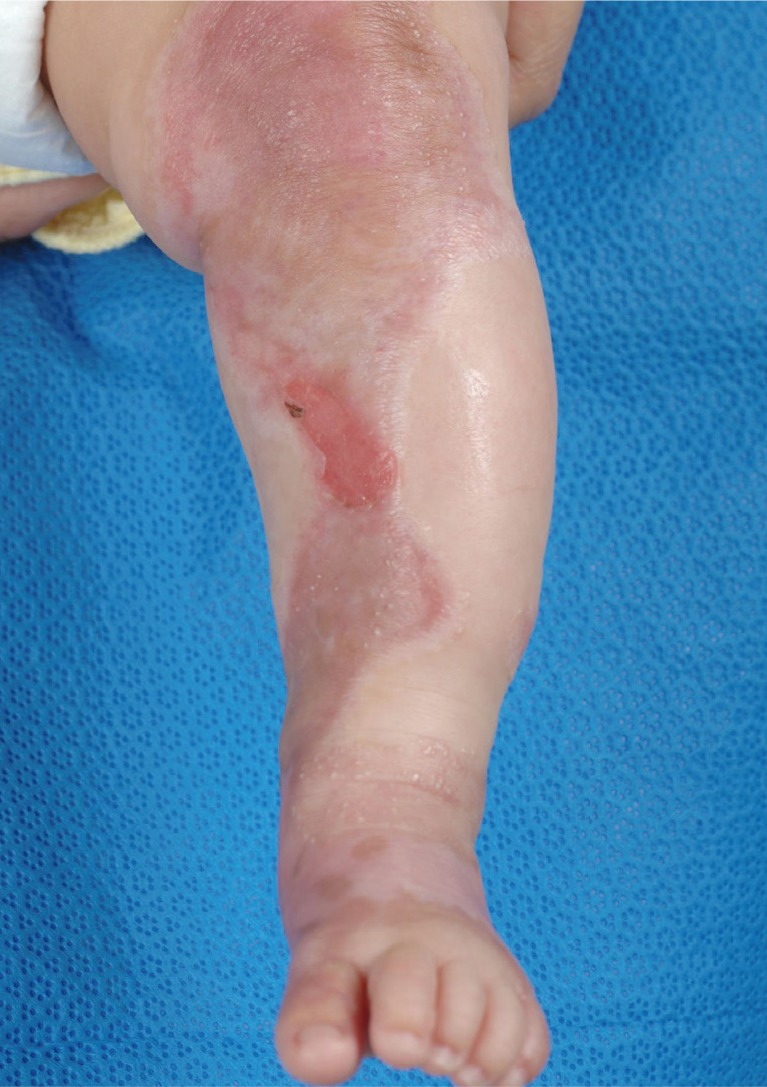
Postreatment 3 month photograph.
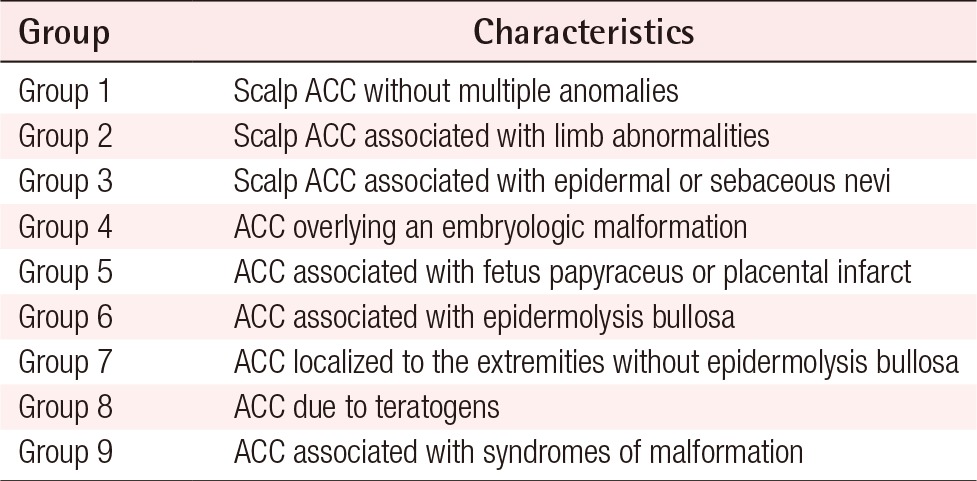
Frieden classification of aplasia cutis congenita (ACC)
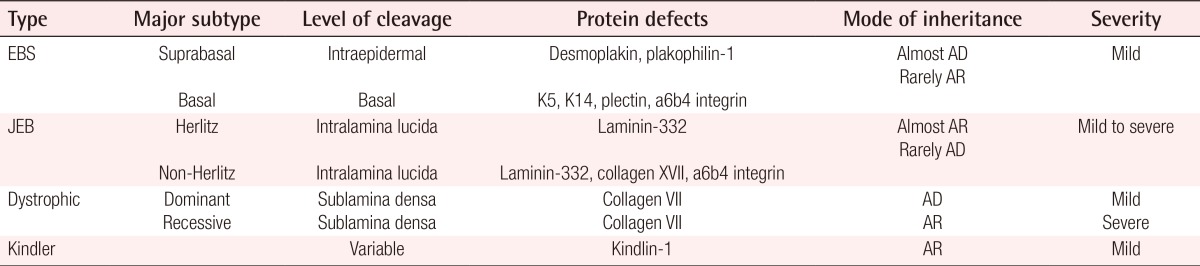
Classification types and major subtypes of EB
Notes
No potential conflict of interest relevant to this article was reported.