Composite Tissue Allotransplantation Immunology
Article information
INTRODUCTION
Composite tissue allotransplantation (CTA) is an option recently introduced for major reconstruction of tissue defects. Since announcements of successful hand, larynx, knee, muscle, nerve, abdominal wall and, most recently, partial face transplantation, CTA has become one of the techniques used by plastic and reconstructive surgeons [1]. Clinical success in CTA is the culmination of progress in two disparate surgical disciplines: replantation and organ transplantation, a close collaboration between plastic and transplant surgeons. This joining of reconstructive and transplant surgery forces the movement of hand and facial tissue allotransplantation into the clinical arena [2]. Translation to the clinical field has shown that CTA is a viable treatment option for those who have lost extremities and suffered large tissue defects [3-5].
As with other allografts, CTA can undergo immune-mediated rejection. When compared with solid organ transplants, composite tissue allografts are histologically heterogeneous, composed of different tissue types (e.g., skin, muscle, bone, bone marrow, lymph nodes, nerve, and tendon), and express different immunogenicity of transplanted elements [6]. Currently, the most important issue for routine application of CTA to clinical practice is the need for lifelong immunosuppression [7]. The immunosuppression medications used to prevent tissue rejection in CTA are the same as those used in tens of thousands of solid organ transplant recipients. The toxicity of chronic, nonspecific immunosuppression remains a major limitation to the widespread availability of CTA and is associated with opportunistic infections, nephrotoxicity, end-organ damage, and an increased rate of malignancy [6]. Because composite tissue allograft transplantations are not life-saving procedures, much attention has been devoted to the issue of minimizing or withdrawing immunosuppression, and this would represent a significant step forward in this field [8]. Over the past five to six decades, advances in the field of transplant immunology have transformed solid organ transplantation into standard care, with excellent short term results in kidney, heart, lung, liver, and pancreas transplantation. The science of CTA is rooted in progressive thinking and the innovative solutions of plastic surgeons. Development of a tolerance regimen or new less toxic immunosuppressive protocols is essential for future acceptance of CTA [9]. With cautious optimism and healthy critiques, the science of CTA promises a bright future. This paper reviews key terminology, drug combinations, mechanisms of immunosuppression, the risks associated with CTA, and immune tolerance protocols.
IMMUNE REJECTION
Antigen-presenting cells
Antigen-presenting cells are a heterogeneous population of leukocytes with efficient immunostimulatory capacity. They are present in the skin, lymph nodes, spleen, and thymus and within or underneath most mucosal epithelia. In skin allograft or CTA, mature dendritic cells play the role of antigen presenting cells. Secondary lymphoid organs like lymph nodes are the sites where primary immune responses develop, and they also drain the site where antigens are deposited (e.g., skin, lung, intestine, blood). Major histocompatibility complex (MHC) antigens are primarily responsible for rejection of genetically different tissues and these molecules are not equally distributed in all cells of the body. MHC class I molecules are constitutively expressed on the surface of most nucleated cells in the body, whereas MHC class II molecules are restricted to the professional antigen-presenting cells, B cells, activated T cells, and vascular endothelial cells [10]. The expression of MHC molecules in cells is controlled by various cytokines and may be upregulated by interferon and tumor necrosis factor (TNF), which are powerful inducers of MHC expression in many cell types. MHC class II molecules are targets for rejection because they bind antigen into their peptide-binding sites and present this antigen to Treg (T) cells [11]. The antigen-presenting cells activating rejection can come from either the donor or the recipient. Allograft rejection may occur by means of two distinct pathways: the direct and indirect presentation mechanisms of allorecognition. Donor antigen-presenting cells migrate out of the allograft to the draining lymph nodes and spleen and stimulate recipient T cells directly. In contrast, indirect recognition requires that the recipient antigen-presenting cells process the donor MHC antigens that have been continuously shed from the graft before presenting them to the recipient T cells in a self-restricted manner. The MHC-T cell receptor (TCR) cross reaction plays an important role in the indirect pathway. It has been suggested that the direct pathway predominates during early acute rejection and that the indirect pathway provides a continuous supply of alloantigen responsible for chronic rejection [12].
T lymphocytes
The thymus is a primary site of T lymphopoiesis and plays an important role as the primary maturational and educational center for developing T lymphocytes. T lymphocyte precursors acquire the ability to recognize antigens through the development of specific surface receptors. The definitive T cell lineage marker is the TCR. Based on the TCR marker, T cells are subdivided into two different types: αβ T cells and γδ T cells. The αβ T cells are responsible for most immune responses, whereas γδ T cells usually respond to antigenic challenges such as viral and bacterial infections and antitumor immunity [13]. Mature αβ T cells are subdivided into two distinct populations that differ in their primary function: a subset that carries the CD4 molecule and mainly "helps" or "induces" immune responses, and a subset that carries the CD8 molecule and is predominantly cytotoxic. Organs and composite tissue allografts can be rejected when the graft contains any antigen that is not present in the recipient. After transplantation, foreign MHC molecules can directly activate T cells. Rejection responses have a molecular basis in TCR/MHC interaction. For T cell activation, a sequence of three signals is necessary. The first step in activation of T cells is accomplished by means of T cell antigen receptors through antigen recognition of donor-derived peptides in association with the MHC antigens expressed on the graft (signal 1). The second step of T cell activation is delivery of co-stimulatory signals through the binding of T cell molecules such as CD28 to its ligands, CD80 or CD86, expressed on donor antigen-presenting cells (signal 2). Antigen presentation to the T cell antigen receptor without the second signal results in clonal anergy. After the co-stimulatory signal is completed, the T cells are able to secrete interleukin-2, which interacts with its T cell antigen receptor and generates a third signal in T cell activation, which leads to T cell differentiation (signal 3) [14]. Activated T cells, mainly CD4 cells, reject transplanted grafts by means of an immunologic mechanism, including the release of proinflammatory cytokines.
Histologic classification
The diagnosis of rejection in CTA is currently evolving. Although most CTA recipients have had acute rejection episodes as defined by graft infiltrating cells, no universally accepted criteria for CTA rejection reporting has been established. Histopathology plays a key role in diagnosis of rejection, understanding the physiopathology of rejection, and facilitating management. Standardization is necessary for reporting clinical results and establishing objective end points for clinical trials. A dispersed and unstandardized development of CTA would present a major barrier for a collaborative relationship in clinical CTA. The recommendations for slide preparation are hematoxylin and eosin (H&E) and periodic acid Schiff (PAS) stains. Immunohistochemical stains are also recognized as potentially important and are thus recommended "as needed" based on H&E findings and/or for research purposes. These include but are not limited to CD3, CD4, CD8, CD19, CD20, and CD68, as well as HLA-DR, CMV, and C4d [15]. There is an evolving consensus that not all graft infiltrating cells are bad. Recent findings have investigated the nature of the cellular infiltrates. Notably, CD4/CD25/Foxp3/CD127 regulatory T cells comprise most of these graft-infiltrating cells in the skin up to 6 years after transplantation and may be beneficial [16]. Future directions include the characterization of the infiltrating cells and their function, the study of accommodation, chronic injury and AMR, the utility of molecular studies, and the inflammatory response in this complex transplant.
Rejection responses
Genetic disparities between the donor and recipient cause the recipient's immune system to recognize grafted antigens as foreign and can destroy grafted tissue. CTA represents diverse tissue components with varying antigenicity, but the rejection response is assumed to be mediated by mechanisms similar to those of solid organ transplantation. In human hand transplant recipients, acute rejection rates were recently reported to be 67% at 1 year [17]. These high acute rejection rates may be explained, in part, by the greater immunogenicity of skin tissue. Despite the relatively high acute rejection rates observed in composite tissue allotransplant recipients, survival rates have been high and all acute rejection episodes were successfully reversed regardless of the anti-rejection therapy used. In solid organ transplantation, high acute rejection rates are often associated with high incidence of chronic rejection and low organ survival rates. Chronic rejection is the most important cause of late graft loss in solid organ transplantation. However, this has not been the experience with composite tissue [18].
Hyperacute rejection
Hyperacute rejection develops immediately, within a few minutes after transplantation, and is an acute cause of graft function failure. Hyperacute rejection is the classic example of the antibody-mediated rejection process and the most important factor in hyperacute rejection is the presence of sensitized antibodies against MHC antigens. Anti-MHC antibodies may be present in the recipient's circulation because of prior blood transfusions, rejection of previous transplants, or multiple pregnancies [19]. In addition, the presence of antibodies against the ABO blood group system can induce hyperacute rejection [20]. Most of the damage that occurs during hyperacute rejection affects the endothelial cells of capillary blood vessels and small arterioles. Immunologic complexes on the vessel endothelial cells activate the complement and coagulation cascade, which eventually leads to damage of the vessel endothelium and ultimately to graft dysfunction [21]. Development of accelerated rejection takes place acutely between 24 hours and 5 days after transplantation; however, this is an infrequent cause of rejection and considered a classic humoral mediated response, which precedes the T cell-dependent mechanism [22].
Acute cell-mediated rejection
Acute rejection, observed in all cases of CTA, is attributable to the primary activation of T cells and the consequent triggering of various effector mechanisms. It occurs within a few days to a few weeks after transplantation and is usually completed within 1 to 6 months [3]. The cellular infiltrate can be mixed and is not limited to lymphocytes. Vasculitis, dermatitis, myositis, and perineural involvement could be confirmed through the evaluation of 4-mm skin punch biopsy specimens taken from the most reddened and/or indurated but apparently viable area of involved skin [23]. Because there is insufficient data to absolutely exclude nonimmune conditions from a particular CTA biopsy, a descriptive observation is currently the appropriate format for reporting findings. Our understanding of rejection in CTA is far from complete [24]. The questions that need to be answered include whether the involvement of different structures (adnexae, epidermis, vessels, etc.) signifies differences regarding outcome, or whether sampling induces diagnostic bias. There is thus a need for developing a new classification that combines both histologic and clinical features (Fig. 1).
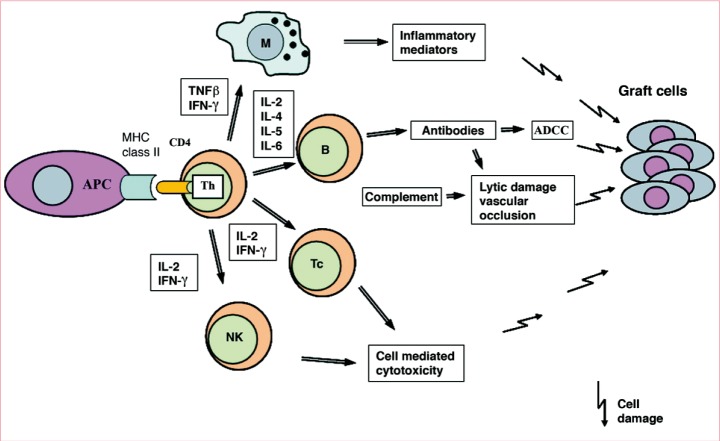
Antibody-mediated rejection
There is not enough information to draw any conclusions regarding Antibody-mediated rejection (AMR), but severe vascular rejection in solid organ allograft recipients has been attributed to a humoral mechanism of the alloantibody-mediated syndrome as a consequence of alloantibody production against donor human histocompatibility antigens. In clinical practice, early acute humoral rejection of solid organ transplants often occurs as severe refractory graft dysfunction, which is resistant to high doses of steroids and does not respond to antilymphocyte antibody therapy [25]. In preclinical observations in a model of nonhuman primates, alloantibodies were detectable after limb allograft rejection. These include the presence of C4d deposition and its relationship with donor-HLA-specific antibodies as well as the presence of vasculitis, neutrophilic margination, thrombi, and necrosis. A complete history including patient sensitization (e.g., cross-match results, transfusions, pregnancies, and previous allografts) as well as the presence or absence of autoantibodies and T- and B-cell crossmatch is to be performed before transplantation [26]. To clarify the role of alloantibodies in composite tissue allograft rejection, future studies are needed.
Chronic rejection
Chronic allograft destruction is a common characteristic of all solid organ and composite tissue allograft transplants and usually occurs within 1 year or later after transplantation [27]. The nature of composite tissue allografts may make them more vulnerable to chronic rejection than solid organ transplants, but the chronic rejection process is still not well understood in composite tissue allograft transplants. Chronic rejection in CTA could be said to be the result of graft injury by immunologic and nonimmunologic factors [28]. In solid organ transplants, chronic rejection leads to a gradual attrition in organ function so that by 10 years post-surgery only 50% of renal allografts are still functional [29]. The frequency of chronic rejection is correlated with several risk factors, including acute rejection episodes, recipient sensitization, insufficient MHC-matched antigens, recipient age and race, inadequate immunosuppression, hypertension, cytomegalovirus infection/reactivation, prolonged ischemic time of the graft before transplantation, smoking, and hyperlipidemia. The hallmark feature of chronic graft rejection is injury to the vessels' endothelium. Initial vascular injury initiates an inflammatory cascade that leads to allograft atherosclerosis and graft fibrosis. Luminal obliteration caused by blockage of the blood vessels occurs because of the proliferation of smooth muscle cells that have migrated from the vessel wall and deposited matrix proteins. Ultimately, fibrosis leads to allograft dysfunction [30].
The fact that organs from living donors are superior to those of cadaveric donors suggests that brain death can influence graft quality and can cause rapid swings in blood pressure, hypotension, coagulopathy, pulmonary changes, hypothermia, and electrolyte abnormalities [31,32]. Organ removal, storage, total ischemia time and engraftment may increase the immunogenicity of allografts by upregulating MHC antigens and activating the cytokine adhesion molecule cascade, which in turn leads to leukocyte infiltration and, ultimately, obliterative vasculopathy and fibrosis [33,34]. Coping with allograft rejection may be much more difficult in CTA than in other forms of transplantation. If a renal or cardiac allograft is rejected, then a retransplantation can be performed and function can be immediately regained. If 50% of a kidney's nephrons are lost, that kidney will still be able to fulfill its functional role of maintaining a normal serum creatinine because kidneys are homogeneous structures. A face transplant, for example, where there has been extensive resection of the recipient tissue at the time of transplantation, may be impossible to reconstruct if rejected.
IMMUNOSUPPRESSIVE AGENTS
Pharmacologic treatment with immunosuppressive drugs will always be the main line of defense against alloimmune reactions initiated by allotransplantation. Current studies are focused on immunosuppressive agents that effectively induce allograft acceptance, with minimal toxic side effects [35]. In clinical practice, these drugs are administered immediately after transplanting the organ or tissues (induction therapy) and regularly thereafter 'for life' (maintenance therapy) and in response to rejection episodes (treatment or rescue therapy). In contrast to solid organ transplants, and because of the different degree of antigenicity of the composite tissue allograft components, the rejection process is mediated by different cellular and humoral mechanisms [36]. As the most immunogenic component of the composite tissue allograft, skin therefore requires a higher level of immunosuppression in comparison with solid organ transplants. Clinical experience with immunosuppressive protocols in composite tissue allografts including hand transplants, abdominal wall transplants, and very recently a partial face transplant, have been based on the protocols used in solid organ transplantation (Fig. 2) [37].
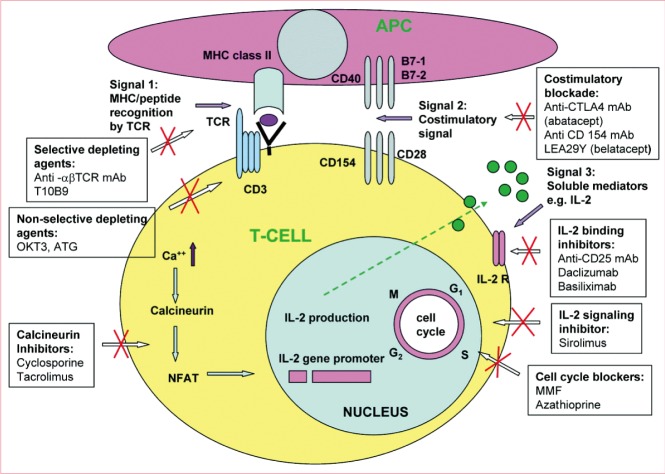
T cell activation and site of action of immunosuppressive drugs. [alpha][beta]-TCR mAb, [alpha][beta]-T cell receptor monoclonal antibody; APC, antigen-presenting cells; ATG, antithymocyte globulin; IL-2, interleukin-2; IL-2R, interleukin-2 receptor; mAb, monoclonal antibody; MHC, major histocompatibility complex antigen; MMF, mycophenolate mofetil; NAFT, nuclear factor of activated T cells; TCR, T cell receptor (From Siemionow and Klimczak [37], Plast Reconstr Surg 2009;123:7e-17e, with permission from American Society of Plastic Surgeons).
Induction therapy
The goal of induction therapy is to silence the immune system so that it becomes unable to initiate an immunologic response that leads to acute rejection. Globally, induction therapy decreases the severity of the first rejection and delays the time to first rejection while allowing time immediately post-transplant to achieve target immunosuppressive levels of the maintenance agents. The four primary drugs currently used clinically for induction therapy in solid organ transplants are: polyclonal anti-thymocyte globulins (ATG); anti-interleukin-2 (IL-2) receptor monoclonal antibodies (daclizumab and basiliximab); Campath-1H and anti-CD3 monoclonal antibodies [38]. In addition to these drugs, donor bone marrow infusion has also been attempted for induction therapy. In an attempt to induce tolerance through microchimerism, a team in Amiens, France transplanted donor-derived bone marrow along with facial tissues. While this approach has been reported to induce tolerance in an animal model, its effectiveness in humans remains mixed. In addition, induction agents have significant costs associated with their use.
Maintenance therapy
The goal of maintenance therapy is to reduce the immune system's ability to recognize and reject the foreign organ or tissue, while limiting toxicity. As the patient progresses further post-transplant, the risk of rejection is reduced and the immunosuppressive regimen is tailored to the individual patient to provide lifelong suppression of the immune system with minimal toxicity. In most cases, each of the drugs used inhibits the immune system at different sites by different mechanisms, which leads to very powerful immunosuppressive effects. This makes it possible to administer low doses of each individual drug and thus reduce the drug-related toxicity. The primary drug combination used for maintenance therapy in CTA is tacrolimus (FK506), mycophenolate mofetil (MMF), and corticosteroids. Due to its effectiveness in suppressing skin rejection and relatively low toxic side effects, tacrolimus-based combination therapy has become the immunotherapy of choice for hand and facial tissue allotransplantation.
The introduction of tacrolimus (FK506) in 1992 led to a decrease in 1-year acute rejection rates from the previous rate of 50% with cyclosporine-based immunosuppression to approximately 30%. Tacrolimus is a macrolide antibiotic, derived from the soil fungus Streptomyces tsukubaensis that prevents T cell activation and suppresses B-cell activation and like cyclosporine is a calcineurin inhibitor [39]. In vitro, tacrolimus has been shown to be 100 times more potent than cyclosporine. It is of interest that tacrolimus has been shown to promote nerve regeneration in small animal models after nerve injury. These effects seem to be related to actions of multiple neuro-immunophilin ligands and may be of particular use in instances such as hand and facial CTA where motor and sensory function is crucial for overall function [39]. In fact this effect of promoting nerve regeneration is thought to be responsible for the 'better than expected' early functional outcomes reported in the clinical hand and facial tissue allotransplants performed. Topical tacrolimus ointment is also used in the clinical setting to treat various skin conditions such as atopic dermatitis, psoriasis, and pyoderma gangrenosum in the form of a topical immunosuppressant.
The introduction of MMF in 1995 resulted in the lowering of acute rejection rates. When combined with tacrolimus and corticosteroid, MMF enabled 1-year acute rejection rates below 20%. MMF is an antiproliferative immunosuppressive drug and treatment with mycophenolate mofetil causes death by apoptosis in a large proportion of activated T cells by inhibiting DNA synthesis and suppressing antibody formation by B cells [40]. Corticosteroids are cytokine gene expression blockers and, along with adrenal glucocorticoids, are the most commonly used immunosuppression drugs. Prednisolone, the prototype in this class, is analogous to the major endogenous corticosteroid. Its actions are mediated by subcellular hormone receptors that form steroid receptor complexes, bind to DNA, and affect the expression of genes driving protein synthesis and cellular processes. It is the first line of treatment in acute rejection episodes. In hand and facial tissue allotransplantation, topical corticosteroids and topical tacrolimus have also been used successfully.
However, when they are unsuccessful, the same powerful antibody-based therapy used in induction is usually started. Rejection episodes have also been successfully treated with high-dose tacrolimus and sirolimus (rapamycin). Sirolimus provides immunosuppression by affecting the G1 phase of the cell cycle and blocking the second signals of activation delivered by interleukin-2, interleukin-4, and interleukin-6 to T cells [41,42]. To minimize the side effects of immunosuppressive protocols, a combination of immunosuppressive drugs is often used for maintenance therapy. Combination therapy has a more powerful immunosuppressive effect than monotherapy because each drug inhibits the immunologic response using a different mechanism.
Risks associated with immunosuppression in CTA
Great advances have been made in immunosuppression over the last 30 years. However, even modern immunosuppressive drugs can be deleterious to a patient's health. Immunosuppressive drugs broadly suppress the immune system, and their use is associated with an increased risk of adverse side effects. The side effects from modern immunosuppressants can be divided into three categories: drug toxicity, opportunistic infections, and finally, malignancies.
Calcineurin inhibitors such as cyclosporine and tacrolimus are nephrotoxic. An elevated risk of hypertension, hyperglycemia, and hyperlipidemia is observed during cyclosporine therapy. Tacrolimus may generate neurotoxic and post-transplant diabetic complications. Cyclosporine is nephrotoxic, can induce hypertension, hyperglycemia, hyperlipidemia, and also gastro-intestinal complications [43]. Though the incidence of these complications varies, nephrotoxicity of some form has been reported in up to 70% of patients. MMF can be a cause of gastrointestinal upset and may induce leukopenia; however, it possesses promising antitumor properties. Steroids are associated with serious complications such as hypertension, diabetes, weight gain, osteoporosis, avascular bone necrosis, gastrointestinal lesions, and poor wound healing [44]. The relatively new immunosuppressant sirolimus is recommended to minimize the risk of post-transplant malignancies, but it has its own side effects, such as hyperlipidemia and the risk of leukopenia. Steroids are associated with a wide variety of problems, in particular delayed wound healing, diabetes, and gastrointestinal perforations.
Eighty percent of solid organ transplant recipients develop some form of infection and 40% of post-transplant deaths are due to infective causes. Of all infections, 55% are bacterial, 30% viral, and 15% fungal. The most serious complication of chronic immunosuppression is the risk of malignancy, which may be an effect of prolonged calcineurin inhibitors and antimetabolite therapy. The incidence of malignancy in transplant patients ranges between 4% and 20%, depending on the immunosuppressive regimen. Most of these malignancies are skin cancers that can be dealt with by surgery but 20% are nonskin malignancies [43]. There is a another specific problem that can be caused by the combination of immunosuppression and organ transplantation; graft-versus-host disease (GvHD). This is a situation where the cells from the transplanted organ, in particular donor T lymphocytes, migrate out of the allograft and attack the host tissue. Common sites affected include the hematopoietic tissues, leading to neutropenia, as well as the gastro-intestinal tract and the skin [44]. The incidence of GvHD is related to the type of tissue transplanted, but any tissue that is rich in donor lymphocytes has the potential to cause GvHD. This may be particularly problematic in a composite tissue allograft that contains a bone marrow compartment, such as a hand transplant, as this is a repository of donor lymphocytes. The concern that the bone marrow in a hand allograft could cause GvHD has led some groups to irradiate the limb prior to transplantation or even to physically scoop out the bone marrow from the transplanted radius and ulna.
Nonimmunologic risks
Immunosuppressive agents may increase cardiovascular risk by affecting cholesterol levels, triglycerides, blood pressure, renal dysfunction, and post-transplant diabetes mellitus [45]. While post-transplant diabetes mellitus has not been reported, transient hyperglycemia occurred in 50% of the hand transplant recipients, primarily while receiving high corticosteroid doses early after transplantation [46]. Noncompliance was a problem in one of 20 patients, and this could possibly have been avoided had a more careful pre-transplant psychosocial screening assessment been performed [47]. The graft may also be 'lost' if a decision is made to surgically remove a viable graft in the presence of drug toxicity, infection, or malignancy, when saving the patient's life is clearly more important than saving the allotransplant. This situation has not been reported in any of the hand or face transplants performed to date. Accepting risk is common to all forms of reconstructive surgery. The new dilemma that CTA brings is that the risk is not just at the time of surgery but continues as long as the patient is on immunosuppression. Proponents of immunosuppression-based CTA argue that it is the patient's choice as to how much risk they are willing to trade for improved quality of life.
STRATEGIES FOR PREVENTING ALLOGRAFT REJECTION
Challenges
Despite the aforementioned limitations, the current success level of hand transplants have exceeded many people's expectations. However, numerous obstacles must be overcome before reconstructive transplantation can be accepted as a routine technique. All organ transplantation requires some form of screening. The donor and recipient must be of the same blood group to prevent hyper-acute rejection due to preformed antibodies [48]. The other matching issue that should be addressed is the degree of immunological similarity or histocompatibility between the recipient and donor. It has been shown in solid organ transplants that the more the donor and recipient differ immunologically, the higher the incidence of rejection. It is not clear yet whether this amount of matching will have a significant influence on CTA survival. However, any reliance on histocompatibility matching to prevent rejection would seriously limit the number of donor-recipient combinations in CTA and would, therefore, preclude the widespread use of CTA [49].
There are some screening issues that are unique to CTA. Most reconstructive allografts are visible and, therefore, must be cosmetically matched to the recipient. Broadly, this means that the donor and recipient should be of the same sex and ethnicity, and also be of similar age. The amount of cosmetic matching required will depend on the transplant being performed; the matching for a face transplant would have to be more accurate than that for a hand transplant. Conversely, a double hand transplant may not have to be as carefully matched as a single hand transplant [50]. Either way this additional matching required for CTA means that donor-recipient pairing will be much harder than for solid organ transplantation. This may require the initiation of international recipient databases and a high level of collaboration between units if the use of all potential donors is to be maximized.
T cell depletion
Many experimental and clinical observations have indicated that elimination of mature T cells is a critical mechanism of transplantation tolerance [51]. The nonselective depletion of T cells (targeting all T cells, not only alloreactive T cells) using antilymphocyte sera, antibodies directed against CD3, and the CD52-specific monoclonal antibody Campath-H1 are frequently used in clinical practice as induction therapy before transplantation [52]. These reagents may also be used for the prevention and treatment of acute rejection episodes. Recently, selective inhibition of allo-reactive T cells by means of depletion of T cell receptor on allo-reactive T cells has been used in experimental studies and in clinical practice [53].
The transient blockade of various co-stimulation pathways using monoclonal antibody has been used as a therapeutic strategy for preventing both acute and chronic rejection. The co-stimulatory pathway CD28/B7 may be inhibited by use of the monoclonal antibody CTLA4Ig (abatacept), which binds to the CD80 and CD86 receptors present on antigen-presenting cells. A second-generation agent, LEA29Y (belatacept), was recently introduced, and it had a higher affinity for CD80 and CD86 molecules than CTLA4Ig and was found to be more effective than CTLA4Ig when used in initial primate studies [54]. In a recent clinical trial in renal transplantation using belatacept, a selective co-stimulatory blocker, as a primary maintenance immunosuppressant, the outcomes were significantly improved. Belatacept appeared to preserve renal function and reduced the rate of chronic allograft nephropathy compared with cyclosporine-treated patients at 1 year after transplantation [55].
Donor bone marrow transplantation and chimerism
From a historical perspective, it is well known that transplanted organs contain large numbers of cells of bone marrow origin known as passenger leukocytes. These cells may migrate from transplanted tissues and colonize lymphoid and nonlymphoid organs of recipients, and the persistence of donor-origin cells within the recipient compartment is known as chimerism [56]. The load of donor leukocytes can be increased in recipients by infusion of donor bone marrow cells. One of the best-studied approaches for establishing tolerance is hematopoietic stem cells (HSCs). Macrochimerism usually occurs when bone marrow (BM) is transplanted to a conditioned recipient. Donor hematopoietic cells can develop two types of macrochimerism. The first is full chimerism, in which the recipient immune system is destroyed by myeloablation and replaced fully (donor cells 100 percent) by donor hematopoietic cells. The second is mixed chimerism, induced after nonmyeloablative host conditioning when donor and recipient hematopoietic cells coexist within the recipient (donor cells 1-100 percent). In this mixed chimerism, the donor and recipient hematopoietic systems co-exist [57]. Mixed chimerism is associated with donor-specific transplantation tolerance in vivo and in vitro and has been shown to effectively induce donor-specific tolerance to a variety of allografts [58]. In humans, BMT-induced mixed chimerism has been shown to confer acceptance of donor-specific skin and kidney allografts without long-term immunosuppression [59]. An additional advantage is that mixed chimerism prevents chronic rejection and is associated with a lower incidence and severity of GvHD and can be induced through nonmyeloablative conditioning [60]. Therefore, mixed chimerism represents a superior approach to the induction of durable transplantation tolerance. The third type of chimerism is defined as microchimerism, which arises as a result of migration of passenger leukocytes from a transplanted allograft into an unconditioned recipient [61]. Passenger leukocytes from the transplanted allograft interact with the recipient leukocytes and are hypothesized to lead to clonal exhaustion, resulting in donor-specific tolerance. In microchimerism, donor pluripotent hematopoietic stem cells do not engraft, but alternatively, hematopoietic-derived cells from the donor organ are produced and migrate systemically. Consequently, not all stem-cell-derived lineages are produced, and very low levels of donor cells are found in the recipient's blood [62]. The phenomenon usually occurs spontaneously after organ transplantation, and donor-origin cells represent less than 1 percent. Microchimerism has been demonstrated in liver and kidney transplant recipients and can be detected by polymerase chain reaction, immunocytochemistry, or other highly sensitive techniques. In human hand allograft recipients, the donor bone marrow is transplanted within its own stromal microenvironment, providing a continuous supply of donor-derived hematolymphopoietic cells, there was transient microchimerism reported [63].
Immune tolerance
Tolerance is defined as a state of donor-specific hyporesponsiveness in the recipient in the absence of immunosuppression. Tolerance to alloantigens is one of the most sought after goals in the transplantation field. For true antigen-specific tolerance, the immune system must not respond to a graft in the absence of ongoing immunosuppression but maintain its ability to respond to other antigens, including viruses and bacteria [64]. Although the main goal in organ transplantation is to develop complete stable tolerance to prevent immunosuppression-related side effects, this goal is difficult to achieve in all organ recipients because of the heterogeneity of donor-recipient combinations, immune status, and the severity of underlying disease [65]. At the present time, the definition of tolerance (normal graft function, normal histology, and withdrawal off all immunosuppression) is an incomplete one and presents a difficult challenge in clinical practice [66].
To maintain self-tolerance and defend the ability to maintain immunity against pathogens, a highly balanced mechanism has evolved for generation of antigen-specific effector cell responses and antigen-specific tolerance induction. Immunologic self-tolerance under physiologic conditions can be defined as a stable state in which the immune system does not react destructively against self-molecules, cells, or tissues. Lack or loss of self-tolerance is likely to result in autoimmune responses, and cellular and tissue damage, and in consequence, can lead to the clinical onset of autoimmune disease [67]. Interactions between antigen-presenting cells and lymphocytes are critical for self-tolerance, and these are known to take place in the thymus (central tolerance) and peripheral lymphoid tissues (peripheral tolerance).
The thymus plays a major role in tolerance through its thymocytes, which recognize self-antigens, and destroy them in the process of negative selection. T cell development in the thymus depends on interaction between T cell receptor and self-peptides bound to MHC molecules expressed on thymic antigen-presenting cells and thymic stromal cells [68]. The mechanisms of positive and negative selection in the thymus are key to the shaping of a self-tolerant T cell repertoire, especially in early life during the maturation of the immune system [69]. In the thymus, developing lymphocytes with no marked reactivity against self-peptides are positively selected in the thymic cortex and enter the circulation as mature lymphocytes. In contrast, developing lymphocytes with marked reactivity against self-peptides undergo negative selection (clonal deletion) in the thymic medulla [70].
Some potentially self-reactive T lymphocytes may escape from the thymus into the periphery. Peripheral tolerance mechanisms are indeed operative in extrathymic lymphoid tissues and include elimination of post-thymic self-reactive T cells, anergy, antigen ignorance, apoptosis, exhaustion, and active suppression by regulatory T cells [71]. Anergy is the state in which T cells are alive but incapable of responding to antigen stimulation. Anergy may be induced by lack of costimulatory signals and, in consequence, this process may lead to apoptosis [72]. Ignorance may occur when donor antigens (e.g., from the graft) are not delivered to the recipient lymphoid system or recipient lymphocytes are unable to recognize donor antigen [73]. The role of regulatory T cells in tolerance is introduced later in the "Regulatory T Cells" section.
Functional tolerance
A functional immune deficit prevents the allograft immune response because of the blockade of cellular activation, proliferation, and differentiation. This operationally tolerant state may be achieved using a variety of mechanisms, such as partial clonal deletion, anergy, cytokine pattern alterations, and the presence of immunoregulatory cells [74]. This new concept of functional or "prope" (almost) tolerance could be defined as donor-specific hyporesponsiveness under a low dose of immunosuppressive therapy [75]. This partial functionally tolerant state with a minimal baseline nontoxic dose of maintenance therapy is suggested to be clinically more appropriate and could be defined as successful organ transplantation with or without the need for maintenance immunosuppression [76]. In an experimental model, tolerance may be monitored in vivo by donor skin grafting or ex vivo by a mixed lymphocyte reaction assay. However, at present, there are no specific in vivo or ex vivo methods available to objectively assess the level of tolerance after withdrawal of immunosuppression [77]. The currently available method for monitoring the clinically tolerant state most clinically accessible would be the biopsy of the transplanted organ to evaluate tissue architecture.
Tolerogenic properties of immune cells
Interactions regulating the dynamic balance between immunity and tolerance are still under evaluation. However, much progress has been made in understanding the essential role of tolerogenic dendritic cells and regulatory T cells in maintaining tolerance. Studies have confirmed the mutual dialogue between dendritic cells and regulatory T cells and show that tolerogenic dendritic cells can be used to stimulate populations of regulatory T cells and promote graft acceptance [78]. In the immature state, dendritic cells express low surface levels of MHC antigens and co-stimulatory molecules and may induce tolerance to foreign peptides [79]. An unresponsive state may be achieved by manipulating dendritic cells to inhibit their maturation process. Efficient cooperation between dendritic cells and regulatory T cells was confirmed in a murine model in which immature host dendritic cells were administered and pulsed with donor alloantigen to induce antigen-specific T cell regulation and was associated with heart allograft survival. In mature form, dendritic cells express MHC molecules (both class I and class II required for the presentation of antigen to either helper or killer T cells) and bear a variety of co-stimulatory molecules such as CD40, CD80, and CD86, which are required for T cell activation and induction of T cell immunity [80]. Mature dendritic cells are also efficient at presenting antigens they have captured at the time of the maturation process; they cannot process and present newly encountered antigens. However, it is evident that the same dendritic cells may mediate either immunity or tolerance, depending on the context of interaction with T cells [81].
After revascularization and implantation of solid organs or CTA, pluripotent stem cells and passenger leukocytes may possibly migrate to recipient lymphoid tissues and colonize them and are then largely replaced in the graft by similar cells of recipient origin [82]. The role of passenger leukocytes in the organs was detected up to 30 years after the first transplantation of kidneys, livers, and other organs. The stem cells and passenger leukocytes are one of the governing factors in immunologic responsiveness or unresponsiveness to allografts, and in tolerance [83,84].
Regulatory T cells
One cell population of interest that has emerged recently is the regulatory T cells. The characterization of suppressor/regulatory T cells has concentrated on the T cell population coexpressing CD4 and CD25 surface antigens [85]. Regulatory T cells also produce Foxp3, a transcription factor that is important in the development of T cells and their role in maintaining tolerance. Regulatory T cells secrete IL-10 and transforming growth factor-β, which have been shown to suppress allograft rejection [86]. Previous in vitro studies have shown that when T cells are introduced into a mixed lymphocyte reaction, they suppress donor-directed T cell responses. Regulatory T cells occur naturally in the thymus and represent a functionally distinct subpopulation of T cells. These cells are characterized as a suppressive T cell population that promotes tolerance to self-antigens and foreign antigens. In clinical applications, immunotherapy with regulatory T cells could provide dynamic control of rejection responses and may result in permanent graft survival, without the need for long-term immunosuppression. Regulatory T cells in peripheral blood (CD4/CD25high/Foxp3) have been shown to express both CCR4 and cutaneous lymphocyte Ag [87]. The presence of these functional skin-homing receptors in most of the circulating Treg cell indicates that they home to normal skin. Development of regimens to promote regulatory T cell expansion ex vivo is one of the promising strategies to achieve tolerance. This approach has been used successfully to generate large numbers of regulatory cells capable of preventing autoimmunity and may be extended for the development of transplant tolerance. A comprehensive analysis of phenotype and function of graft-infiltrating cells needs to be performed to elucidate the exact role of Treg in CTA.
CONCLUSIONS
In summary, CTA is rapidly emerging from an experimental model to a standard of care and holds great potential for reconstructive surgery. The technical aspects of CTA are no longer the factors limiting the widespread application of this treatment modality in the clinical setting. Progress made in the field of immunomodulation, namely conditioning protocols and immunosuppressive therapy, has significantly advanced the field of CTA. However, the uniqueness of the tissues in CTA means that there are inherent adverse effects. In this sense, the field of CTA can not only learn from the solid organ transplant community, but perhaps it can teach as well. This review of transplant immunology focuses on the science of CTA and a comprehensive and understandable review of some of the key immunological principles relevant to CTA. It is important that surgeons in both disciplines have a working knowledge of the relevant scientific and technical principles in their respective areas. This includes knowledge of the mechanism of graft rejection and failure as well as the principle of immune reaction, routine regimens, dosages, and toxicities of the immunosuppressive drugs and immune tolerance-inducing strategies. The induction of donor-specific tolerance would open new options for routine application of CTA in plastic and reconstructive surgery.
Plastic surgeons play a central role in treating facially disfigured individuals and will thus lead the development of these new reconstructive treatments. It is hoped that this paper will serve as a reference for the readership to consider and discuss CTA with their colleagues and patients and advocate further scientific studies before the broad application of composite tissue transplants. This should be of interest to practicing plastic surgeons because composite tissue allografts may become, in the near future, a routine part of their practice.
Notes
No potential conflict of interest relevant to this article was reported.
